Simulating a simple 1D experiment with 5 spins:
A3B2 spin system
Pulse Program: zg
Here we will give explicit step-by-step
instructions for simulating a 1D proton spectrum for a 5-spin system
(A3B2) similar to that in e.g. bromoethane.
We assume that you have launched the Virtual NMR
Spectrometer. Next, enter the spin
system. This can be done manually, as
illustrated below, or you can load the bromoethane spin system by pressing the
‘load’ button at the bottom of the main panel, selecting (navigating to) the
subdirectory called lib, and selecting the file bromoeth.vs1.
When loading the spin system, be sure to select ‘spin system’ and not
‘all’.
STEP 1: Define the Spin System:
The spin system selected for this example consists of 5 protons: three A-protons
and two B-protons, coupled with each other by a scalar coupling. In the case of bromoethane,
the A-protons represent the methyl group and B represent the methylene group.
For simplicity, we will set scalar couplings between the three A-spins to 15 Hz,
between the two B-spins to 12 Hz, and those between the A and B spins to 7 Hz.
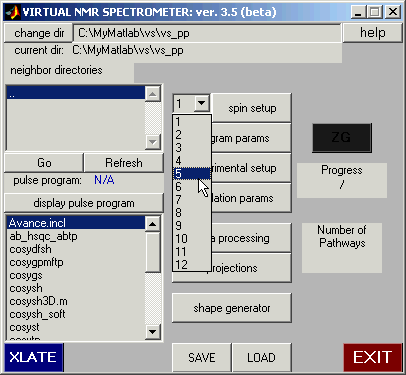
Use the pull-down panel to select 5 spins.
Then click on the ‘spin setup’ button. This brings up the spin setup dialogue. Select
the ‘Graph Mode’ button.
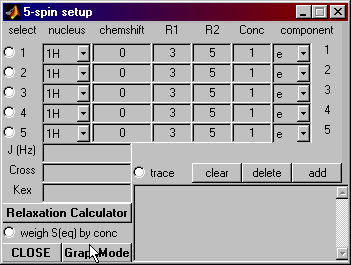
The graph mode is a user-friendly way to input
chemical shifts and couplings. Pressing
that button opens the graph mode window shown below.
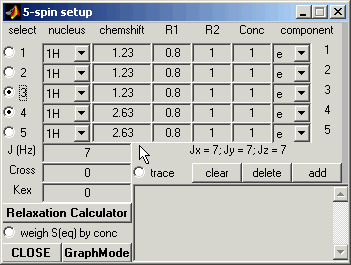
Spin Setup: Graph Mode
The boxes numbered 1 through 5 represent the 5 spins in your spin system.
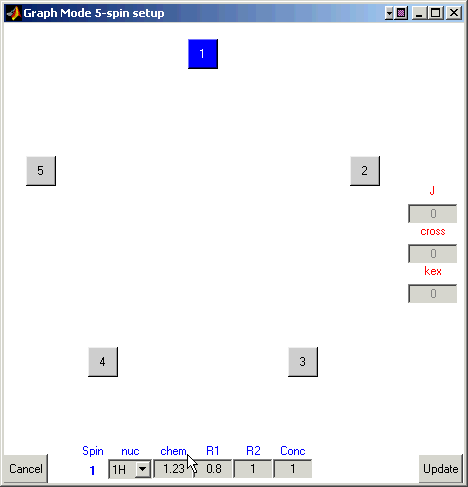
Enter 1.23 for the chemical shift value for the first
nucleus.
Here you can also define the type of the nucleus, its spin-relaxation rates,
and the concentration of the molecular species that this atom belongs to.
For example, enter 0.8 and 1.0 (reciprocal seconds) for R1 and R2, respetively,
-- we will use these values of the relaxation rates for all other spins.
Now let us define resonance frequencies for all
other nuclei. Click on the box for the first atom once to remove the blue
highlight.
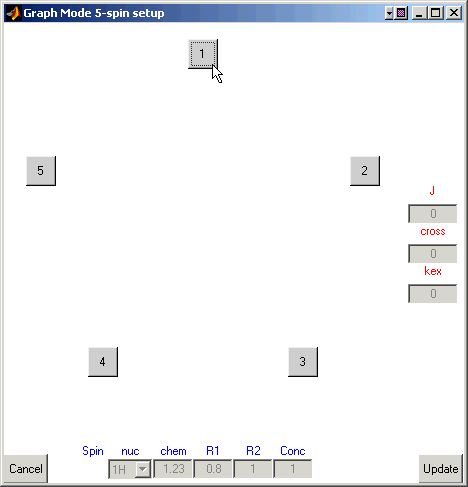
Click on the box for the second atom (it should become blue).
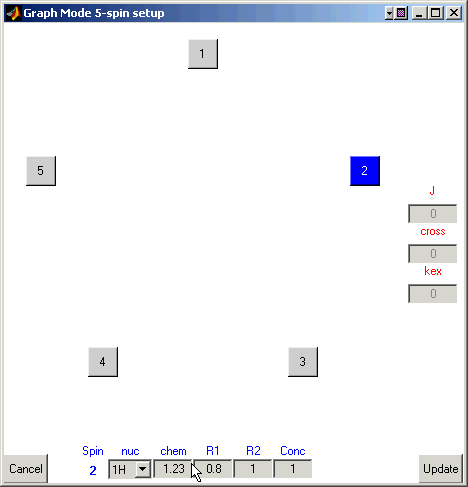
Enter 1.23 for the chemical shift and 0.8 and 1 (reciprocal seconds) for R1 and R2.
Continue entering the chemical shifts for the rest of the atoms. Here’s a table of
the chemical shifts:
|
Atom
|
Chemical Shift
|
|
1
|
1.23
|
|
2
|
1.23
|
|
3
|
1.23
|
|
4
|
2.63
|
|
5
|
2.63
|
Remember to deselect one atom (click it so it
isn’t blue) before moving to the next.
Once all of the chemical shifts have been entered,
define scalar couplings between spins.
Deselect the atoms so that none are highlighted. Now select atom one (highlighting it) and then select atom two.
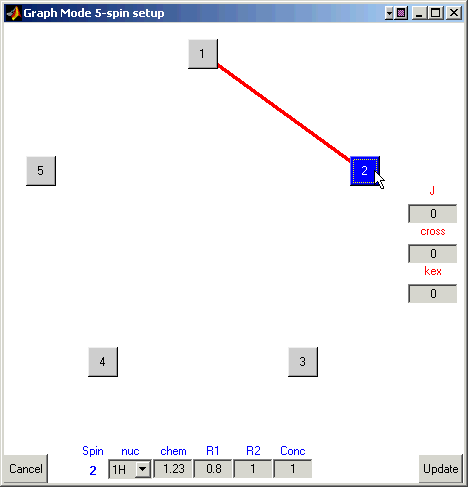
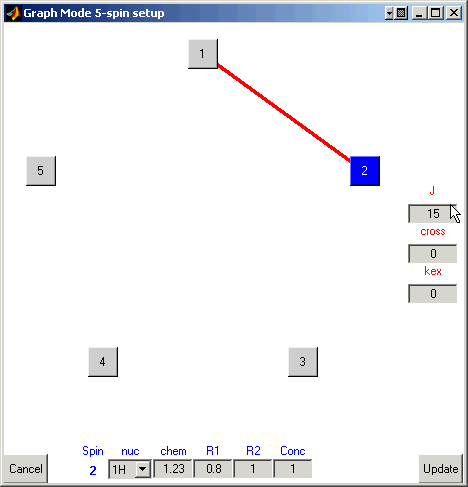
A line will form between them.
Enter a coupling of 15 Hz in the box under ‘J’.
Continue selecting pairs of atoms and input 15 Hz for scalar couplings
between spins 1, 2, and 3, 12 Hz between spins 4 and 5, and 7Hz between these two
groups of spins. Once finished the ‘Graph Mode’ window will look like this:
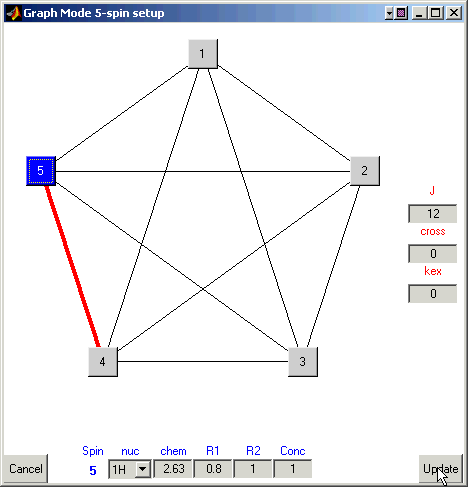
Finally, click on the ‘Update’ button to save/input your data into VNMR. This
takes you back to the ‘spin setup’ window.
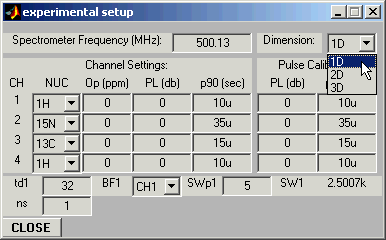
STEP 2: Experimental Setup
Now we are ready to set up experimental conditions for the simulation.
Open Experimental Setup Panel and set experiment dimension to 1D:
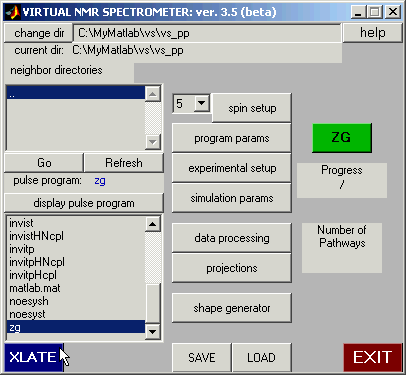
Return to the Main Panel, select pulse sequence zg, and then
click on the XLATE button to translate it. If the translation is successful, the
ZG button will turn green.
Open Program Parameters panel and set pulse sequence parameters: in
this case the only parameter that is not assigned and requires to be set is the
recycling delay d1.
Program Parameters:
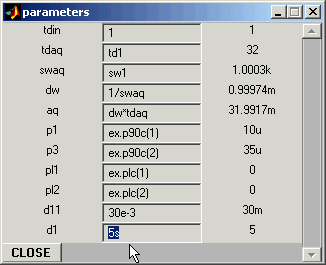
Set d1 to 5s.
Experimental Parameters:
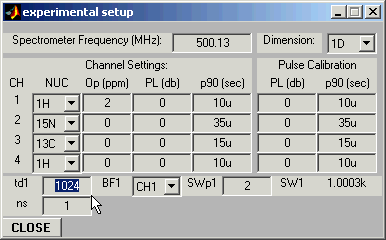
Open Experimental Setup Window and set the carrier position (Op(1)=2
ppm in this example), the number of acquired experimental points you wish to
simulate (td1 = 1024), and the desired spectral width (SWp1=2ppm)
:All relevant entries in the program parameter window will be updated automatically.
STEP 3: Run Simulation
Click on the ZG (green) button -- this will start the simulation.
STEP 4: Data Processing.
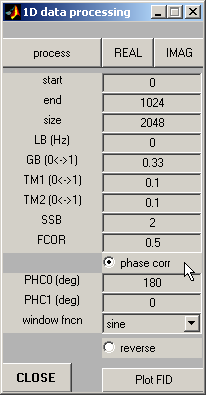
Open Data Processing
window, set zero-order phase correction PHC0=180 and check out the Phase
Correction button.
Click on the Process button to process the simulated FID, and then on
the REAL button to display real part of the resulting spectrum.
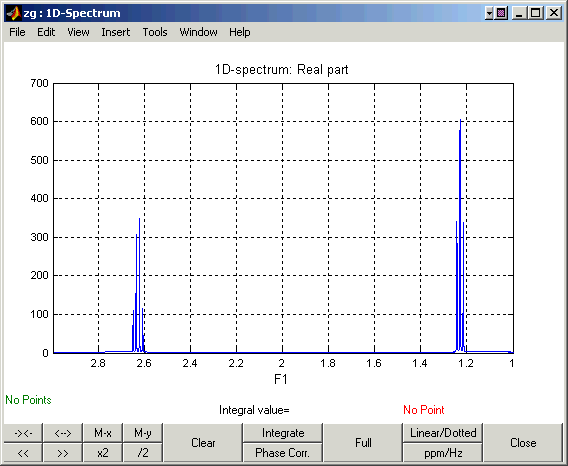
The resulting spectrum should look like this one:
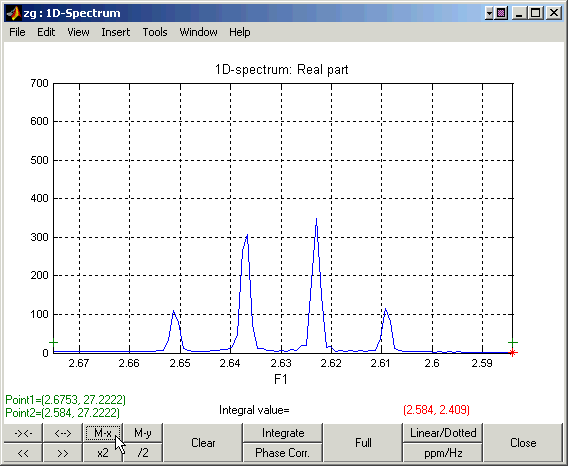
Zooming in on the down-field and up-field regions of the spectrum:
Go Back
Back to Top
Go Back to VNMR Manual
This manual page was designed by David Fushman & Charles Geraghty.
(c) Copyright 2004 by David Fushman, University of Maryland.